|
Register to attend this year's event at the Indiana Statehouse on 2/12/24 from 11a-2p. Click the link to register https://SCDAdvocacyDay2024.eventbrite.com. Help us raise awareness about the barriers that are faced on a daily basis for those in Indiana who have Sickle Cell disease! 
0 Comments
Announcing the 2020 Sickle Cell-abration Video Contest: My Journey, Our Drive! See official rules and guidelines below, and download the contest registration form to enter!
Sickle Cell Video Contest: Official Rules, Guidelines and Entry Form Please read the entire Rules and Guidelines prior to registering and submitting an entry Purpose: To increase understanding of Sickle Cell Disease (SCD) through a personal video production. Three Prizes First place: $100 gift card Second Place: $50 gift card Third Place: $25 gift card (Entries may be shown at Sickle-Cell-Abration, appear on the websites and various social media platforms of Sickle Cell-Abration, IHTC, ISCC and the Martin Center) Eligibility:
Submission Criteria:
Deadlines:
Additional Information
Frequently Asked Questions (FAQs)
For problems, questions and more information, contact Kisha Hampton, Sickle Cell-Abration Event Contest Chair, at 317-871-0011 ext. 366, 317-358-5919 or khampton@ihtc.org Surveillance is finding out how many people have a disease. It is also about finding out how a disease affects people’s lives. The Centers for Disease Control and Prevention (CDC) have a sickle cell surveillance program. It is called the Sickle Cell Data Collection (SCDC) Program. Indiana is working with the CDC on this program.
What is the Goal? The goal of this program is to improve the health of people with sickle cell. The program collects information to improve care for people with sickle cell. For example, the program collects information about where people with sickle cell disease live. It also collects the locations of health care providers. This will tell us how far people with sickle cell disease live from health care. People who make the laws can also use this information. They can look at where the need is for people with sickle cell. It can help them to decide what programs to fund. Where does the information come from? The program collects information about people with sickle cell from many places. These include:
You can learn more about the program by clicking here or by calling the Indiana Hemophilia and Thrombosis Center at 317-871-0000. Thank you to all who joined us for this year's Sickle Cell-abration, Journey Through the Jungle! People with sickle cell disease were joined by their loved ones at the beautiful Hulman River House at the Indianapolis Zoo and White River Gardens. Guests heard from Dr. Simone Uwan, a physician and Sickle Cell Warrior. Guests received signed copies of Dr. Uwan's book and were able to visit the Zoo. It was a beautiful day, and we're already looking forward to next year's event!
   Camp Silver Moon 2019 is in the books, and what a week it was! From the opening campfire to the closing ceremony, every minute was packed full of fun. Laughter from children as young as 7 and as old as 17 echoed through the trees as nearly 50 campers explored the campgrounds this June. Favorite activities included playing ga-ga ball on the beach, rock climbing, and zip lining through the woods. Campers learned about the importance of hydration in sickle cell disease and shared stories of self-care. The most courageous and most compassionate campers were honored with special recognitions at the closing ceremony, and every one of our campers completed a series of sickle cell challenges to earn their Adventure Stick! We are so proud of each of our campers who shone brightly at Camp Silver Moon. Thanks for the memories!
Rare Disease Week is a chance for people from across the country who are affected by rare diseases to come together and share their stories with lawmakers. Believe it or not, sickle cell disease is considered a rare disease in the United States. The term "rare disease" refers to any disease that has fewer than 200,000 sufferers in the U.S. There are about 100,000 people with sickle cell disease in the U.S. Sickle cell advocates from Indiana attended Rare Disease Week 2019 and had the opportunity to talk to Indiana legislators, meet with rare disease advocates from other states, and raise awareness for rare diseases. Advocates attended conferences, receptions, and one on one meetings with members of Congress and their staff. It was a great chance to hone advocacy skills, network, and learn more about the federal lawmaking process!  
January 14, 2019 was a cold and snowy day, but the weather couldn't stop us from holding our 2nd Annual Sickle Cell Advocacy Day at the Indiana State Capitol! Our goal this year was to get a hearing for a bill that would provide funding for services for adults with sickle cell disease. People with sickle cell, their friends and loved ones, and medical and community providers all came together to listen and share stories on what adults with sickle cell experience when they seek medical care or look for a job. Our team was able to talk to many legislators about this important issue, and to encourage them to support Rep. Porter's HB1354, which would provide funding for adults with sickle cell in Indiana. This bill is now in the House Ways and Means (budget) committee, and we are hopeful that soon we will see some real changes for people with sickle cell disease in Indiana! 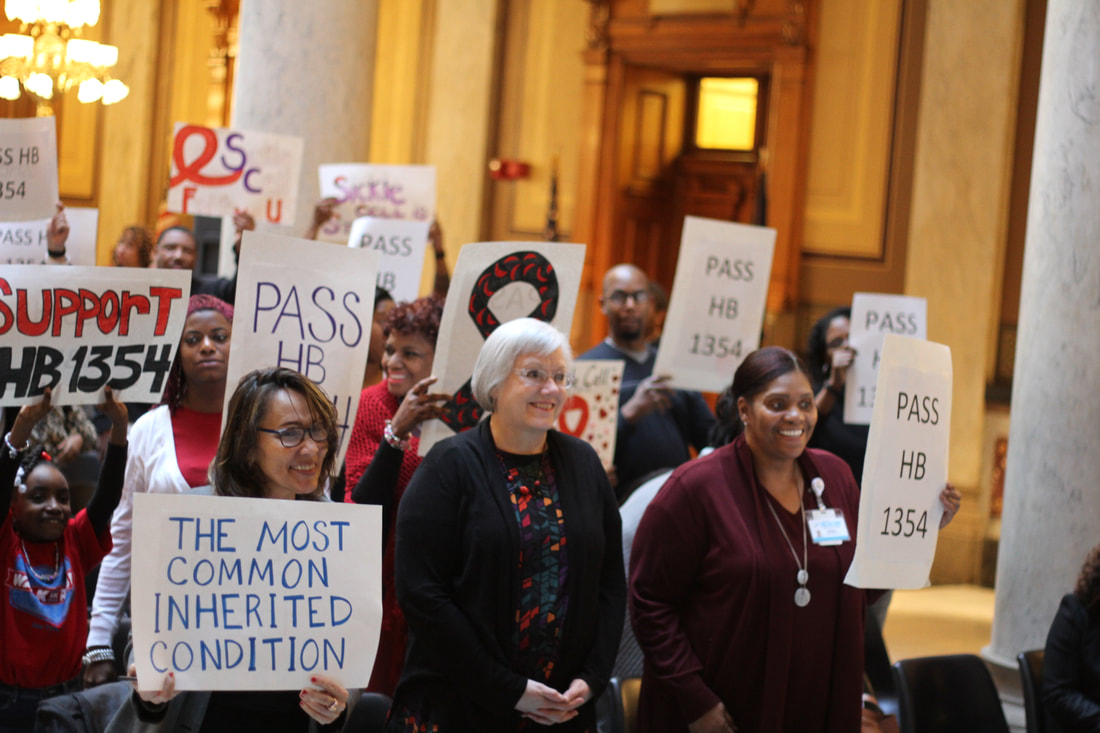 Transitioning from Pediatrics to Adult Care has been one of the most challenging tasks of my life in dealing with my overall health, which also includes dealing with sickle cell. Developmentally, mentally, emotionally, and spiritually, it has without a doubt called for me to “grow up!” in more ways than we even have time to discuss. But, while we’re here, I’ll give you all the low down on my personal experiences recently. I’ll also provide small things we all can do one step at a time. These small things will ensure that the BIG task of becoming a responsible adult living with sickle cell that is mindful of their own strengths and weakness is checked off on our life’s (sometimes long & complicated) list of To-Do’s.
The first major concept I’ve had to learn & accept is that in the adult world, freedom & responsibility go hand in hand. And unfortunately, it’s a part of life us kiddos cannot escape. When I was a child, I wanted so badly to be independent and “on my own” to prove I could handle it all, and now that I’m here…ironically that’s what I got. I am. On. My. Own. But the good news is that once we realize it is OUR responsibility to learn and know what is best for us, it gets clearer and easier for us to ensure we get the best treatment, and quality of life. At that point, freedom is expected and within arms’ reach. Recently, I decided I was too uncomfortable in my safety net of life and needed to stop just flapping my wings while sitting, and actually learn how to FLY. So I set out to use my career as a Registered Nurse in Rehab and my motivation to continue my entrepreneurial vision with ROJOrganics to land a travel nurse contract in North Carolina. On this assignment lasting a minimum of 3 months, I’d have to learn how to begin to fully take control of my life, discover more of who I am, and what I like (or don’t). I hadn’t been hospitalized in almost a year, and not one pain crisis in approximately the same time. I didn’t think sickle cell would be an issue or concern for me looking at all the freedoms I would gain. But goodness, was I wrong. I experienced my first crisis after less than a month of being away from friends, family, healthcare providers—everyone and thing to that I could see face to face in my safety net. I stayed home a few days and cried in a hot-water filled tub, in pain and out of fear, of what was next. “What did I do to make this happen?” “Do I need to go to the hospital?” “Who can I call to get me there?” “How much will my insurance be billed?” “How much time will I need off from work?” “Do I have enough pain medicine?” “Will the ER treat me like I’m a drug addict?” And the list goes on and on. Thankfully, the pain subsided in a couple days and things returned to normal. I signed an extension on my contract after prayer and guidance. But then it happened, AGAIN. Sickle cell silently sneaked into my chest and ribs, showing its ugly face, and forcing me to deal—all while still recovering from the Flu! This time I had to have a different approach, as I spent over a week off from work, dealing with pain, lack of money & lack of providers (the closest sickle cell experts were an hour and half away in a neighboring city). This would take much more time, effort, and resources to solve. I had to realize what was a priority, and do my best to snatch back the control that sickle cell often threatens to take away from me, everyday. And so finally, here’s my important list of To-Dos and resources that make putting on big girl panties and big boy britches possible and least painful in the pediatric to adult transition.
If you’re like me, then seeing and believing that independence not only can happen but also will happen when the desire brings forth effort is a great sigh of relief. I don’t have all the answers to this because I’m still living and learning. Independence looks differently for everyone due to experiences and circumstances, but these are great starting points. Happy living, my transitioning friends! - Jade J. Parker | Daughter of Christ | Sickle Cell Warrior & Advocate | Registered Nurse | Founder & CEO at ROJOrganics | Sister and Friend 2017 was a banner year for sickle cell disease drug therapy!
For almost 20 years, the only FDA approved medication for sickle cell anemia was Hydroxyurea. Hydroxyurea was approved for adults in 1998. There is much evidence for its effectiveness and safety in infants as young as 9 months, and it has been safely used in children as well as adults since 1998. In 2017, the FDA officially approved Hydroxyurea therapy for children with sickle cell anemia--types HBSS and HBS beta zero thalassemia--starting at age 2 years. In 2017 the FDA also approved L-glutamine (Endari), therapy for sickle cell disease. What is L-glutamine? L-glutamine was approved by the FDA for sickle cell disease in July 2017. As of March 2018, it is on the market as brand name Endari. It is approved for children over age 5 years and adults with sickle cell disease. L-glutamine is a natural amino acid that our bodies make or that we receive when we eat meat or plant proteins such as red cabbage, beans and raw spinach. This product has been studied in critically ill patients, patients with allergies and patients with gut problems in the past. You may have even seen L glutamine on the shelves with supplements used by body builders. The Emmaus Company has studied this treatment since the late 90s in sickle cell patients. The L-glutamine is manufactured from the sugar cane plant, so is actually vegan. It is purified to pharmaceutical grade as brand name Endari. This makes it different from the L-glutamine capsules that are on the shelves of the supplement stores as these cannot be guaranteed for purity. How do you take L-glutamine? Endari comes in a powder form. It has to be mixed well in 8oz of a cold drink . It can also be mixed into yogurt and applesauce. Hot drinks cannot be used as they make L-glutamine inactive. It is taken twice a day and the number of packets depends on your weight: A child less than 66 pounds needs 1 packet twice a day. A person 66 to 123 pounds needs 2 packets twice a day. A person over 143 pounds needs 3 packets twice a day. How does it affect sickle cell patients? A study carried out in 31 centers in the United States led to FDA approval. Patients on the study could continue their Hydroxyurea. Patients had fewer pain crises in a year (an average of 3 a year for those on the study, and 4 a year for those not on the study). They had fewer acute chest syndrome complications. The study also showed that patients had fewer hospital visits than those not taking Endari, and that their hospital stays lasted a shorter time. Patients also experienced longer times between their pain crises. What else should I know about L-glutamine? The most common side effects reported with its use were nausea, constipation and cough. Don’t use Endari if you have trouble with liver or kidney function. It can be used with Hydroxyurea if you are already taking this or plan to start this. How does it work? The way L-glutamine works is not yet fully understood. Red blood cells usually live for 120 days but sickle red blood cells are prone to dying quickly, causing anemia. This red blood cell death is called hemolysis. One of the main reasons for hemolysis is damage to the red blood cell membrane. This damage is caused by natural byproducts in our body called reactive oxygen species. Our red cells are protected against this damage by the NAD (nicotinamide adenine dinucleotide) system. L-glutamine helps us build up our NAD and allow better red cell protection. L-glutamine may also make red blood cells less sticky, which could reduce the number of painful crises. How can I learn more about L-glutamine? Speak with your Hematologist about L-glutamine (Endari). Your sickle cell care team will help you decide if you need one or both of the two preventive medications for sickle cell care (Hydroxyurea and Endari). Your Hematologist also knows if your liver and kidney function are healthy enough for you to tolerate Endari. Your insurance will review your records and approve the Endari on a case by case basis with the help of your Hematologist. Promising medications in the pipeline This recent drug approval after a gap of almost 20 years is very promising for our sickle cell community. We have many medications that are in development by several companies. The only way these can be approved for use is with the help of sickle cell patients who participate in clinical trials. We look forward to the day when we have many different medications options out there to help all our child and adult warriors with sickle cell disease live life to the fullest! Angeli Rampersad Pediatric Hematologist Clinical research is booming! There are currently over 270,000 active studies on https://clinicaltrials.gov. As a research coordinator, I want to discuss the need for participating in research initiatives for Sickle Cell Disease (SCD), as well as what is involved.
As of today, there are about 500 studies devoted specifically to Sickle Cell Disease (SCD) which is a higher number than in the past. What does this mean for people with SCD? This means that pharmaceutical companies are not only interested in SCD, but they are actively funding research to find new drugs to treat it. Currently, there are only a couple of drugs approved for use to help prevent most SCD complications. If you cannot use those medications then management of the disease can become more difficult. In order to generate more medication options for SCD, more individuals have to enroll in research studies. It’s not as simple as approving a new drug for use. Research medications must be evaluated on how well they work in humans. Research is heavily regulated and monitored by organizations like the US Food and Drug Administration (FDA) and local Institutional Review Boards (IRBs), so every drug approved for human use, even Ibuprofen and cough syrup, has been tested on a human subject. Enrolling an individual in a research trial requires several steps. First, anyone who wants to enroll in a study has to fit the guidelines for who can participate, otherwise known as eligibility guidelines. Second, the informed consent form (ICF) must be reviewed. The ICF is a document that explains, in layman’s terms, the study procedures, risks/benefits, requirements to participate, and what is hoped to be gained from the study. These documents go into great detail to eliminate questions and concerns about what will take place during the study. The ICF is reviewed, page by page, before consent to participate is obtained. The most important thing to remember is that consent is voluntary, non-binding, and can be withdrawn at any time. After the patient provides consent, he/she must complete a screening process to validate the person’s eligibility and have multiple clinic visits for labs and routine monitoring. In other words, research requires a lot of steps! As a research coordinator, I love what clinical trials provide for medicine. Hopefully, this has sparked interest for others as well. For more information, contact Adrianna, IHTC Research Coordinator, at 317-871-0000.  What is Sickle Cell Disease?
Sickle cell disease is a disease of the hemoglobin. Hemoglobin is what is inside your red blood cells, carrying oxygen all over your body. Most people have what is called Hemoglobin A, which is shaped like a fist and keeps your red blood cells round and squishy. These red blood cells are able to move through your blood vessels easily. People with sickle cell disease have Hemoglobin S. Hemoglobin S is shaped like a rectangle, and it stretches out your red blood cells (see image below). These stretched out cells (also called sickle cells) are rigid, sticky, and fragile. They get stuck in your blood vessels, and can damage your organs. Some people have sickle cell trait. A person with sickle cell trait has Hemoglobin A and Hemoglobin S. Since they have Hemoglobin A, they usually don’t have health problems from the Hemoglobin S. There are other kinds of hemoglobin besides Hemoglobin S and Hemoglobin A. Some people make Hemoglobin C or E, or another unusual type. These types can also be combined with Hemoglobin S to cause different types of sickle cell disease. How do you get sickle cell disease? Sickle cell disease is an inherited disease, which means you get it from your parents. In order for a person to have sickle cell disease, at least one of their parents must have sickle cell trait. Their other parent must have sickle cell trait or another hemoglobin trait. A person with a hemoglobin trait makes two kinds of hemoglobin, but they can only pass down one kind to their child. If two parents both have sickle cell trait (Hemoglobin AS), they can each only pass down one kind of hemoglobin. If one parent passes down Hemoglobin A and the other passes down Hemoglobin S, their baby will have trait. If they both pass down Hemoglobin A, their baby will just make the usual hemoglobin. If they both pass down Hemoglobin S, their baby will have sickle cell disease (Hemoglobin SS). People can get other types of sickle cell disease if one of their parents passes down Hemoglobin S, and the other parent passes down another unusual hemoglobin, like Hemoglobin C. This person would have Hemoglobin SC. There are many different types of hemoglobin. It’s important to know what type you have, and what type your partner has. On January 29, members of the Indiana Sickle Cell Advisory Committee and the Indiana Sickle Cell Consortium participated in Sickle Cell Advocacy Day at the State Capitol. Several IHTC staff members and patients attended. The focus of the day was to educate legislators on the need for funding for programs and services for adults in Indiana with Sickle Cell Disease. Currently, the state funds two programs at IHTC for patients: the Sickle SAFE Program, for ages 0-3, and the SCORE Program, for ages 3-21. IHTC staff members and other consortium members agree that the state needs to do more for adults. The day’s agenda included speeches from several stakeholders in the sickle cell community, including patients, parents of patients, medical professionals, and even Representative Greg Porter, who has long been a sickle cell champion. Several other legislators visited the event or sent their staff members to gather information. We have plans to make 2019 Advocacy Day even bigger and better! Keep watching this space for more information!  A person inherits genes that produce hemoglobin from his/her parents. It is important to identify people with hemoglobin trait so they will be aware of their risk of having children with sickle cell disease. If one parent has sickle cell trait and the other parent has normal hemoglobin, there is a 50 percent chance with each pregnancy that the child will be born with sickle cell trait. If both parents have sickle cell trait or another hemoglobin trait, there is a 25 percent chance with each pregnancy that the child will have sickle cell disease or another hemoglobin disease.
People with sickle cell trait usually do not have any disease symptoms. However, it is possible for a person with sickle cell trait to have complications of the disease under extreme conditions, such as:
I had some familiarity with Sickle Cell Disease (SCD) when my son was born in November 1996. I was told that he had Sickle Cell Trait (SCT), which he inherited from me. SCD became a part of my family December 25, 2002 with the birth of my twin daughters. One of my daughters had the trait and the other had the disease (HbSS). I didn’t know much about the disease at that time, but I would soon become inundated with information through independent learning and education by doctors and others in the sickle cell world.
At that office visit, we received the name of a sickle cell educator, a referral to the hematology clinic at Riley Hospital for Children, and an address/invite to the Martin Center’s sickle cell support group. Our family was armed and ready for battle! We did not know it at the time, but these resources would prove to be extremely valuable to us and ultimately part of our extended family. They were our front-line supporters as my daughter began to experience complications related to SCD, endure hospitalizations, and require extensive care and support. My purpose for sharing this backdrop is to encourage anyone reading this article to make sure that there is proper coordination of care for any sickle cell patient in your life: your child, your patient, your friend, your student, or anyone else. This begins with having the right team on board to support the patient’s needs. My daughter’s team is extensive, beginning with our biological family and spiritual support. Her medical team includes her primary care physician, dentist, and hematologists at the Indiana Hemophilia and Thrombosis Clinic (IHTC). She also has access to wrap-around care provisions at IHTC including Physical Therapy, Dental Hygienist, Phlebotomy, Dietary, and Social Services. Her hematologists collaborate with other service providers including pulmonary care at Riley Hospital, optometry care, and radiology for her annual Transcranial Doppler scans, just to name a few. IHTC is also the bridge between home and school, for educating and collaborating with school nursing staff and teachers to ensure our daughter has the best support possible. As you can see, this team is extensive. The number one best advocate for a child with SCD is the parent or caregiver with whom the child resides. This advocate must always be alert to ensure the care team is aware and ready. Recently, my daughter’s dentist referred her to an endodontist to remove all four of her wisdom teeth. We consulted with the endodontist and scheduled the surgery. Immediately after the office visit, I reached out to three people at IHTC: her hematologist, her primary nursing contact, and our social/educational advocate to advise them of the upcoming surgery and understand their requirements from a sickle cell perspective. I quickly learned that care coordination is critical. Her hematologist reached out to me immediately with questions and copied the dental hygienist on staff. They both followed up with me again, then reached out to the endodontist directly. Timing was great because my daughter had a clinic appointment coming up before the dental surgery. We were able to have an on-site consult with her IHTC care team, and receive resources/instructions for her post-op care. This resulted in a smooth process and recovery. Don’t take anything for granted. Proper care coordination is critical to ensure that those with Sickle Cell Disease have the best available treatment, care, support, and possibility for effective recovery. Jacquelyn Robinson, MSM, M.Ed., PHR, GPHR Parent and advocate for Sickle Cell patients and families; founder of SCACURE Networks, Inc. People with sickle cell disease (SCD) make hemoglobin S. This hemoglobin stretches out the red blood cells (also called “sickling”). Sickled red blood cells have trouble moving through the blood vessels and can cause pain and organ damage. Hydroxyurea helps people with SCD by increasing the amount of Hemoglobin F in the body. Hemoglobin F helps keep red blood cells from sickling, so it can prevent some problems caused by SCD.
Hydroxyurea has been studied for decades to see how it can help people with SCD. There are several benefits that have been discovered. These include:
Hydroxyurea has been used in patients with SCD for over 30 years. Research has shown that hydroxyurea is a safe and effective medication. People with SCD can start taking it as young as 9 months of age. If you have questions about hydroxyurea, please talk to your or your child’s hematologist. Dr. Clark Kramer Merrillville, Indiana September is National Sickle Cell Awareness Month. We honor and hope for all the #SickleCellWarriors who strive and survive and thrive every day.
Did you know the Partners Program hosts CME accredited sickle cell disease (SCD) modules, free to the public? The website, www.PartnersPRN.org, hosts three CME activities covering the issues facing adolescents and young adults (AYAs) with SCD and the phase of transitioning from a pediatric care model to an adult care model, a period associated with significantly increased risk for care discontinuity, acute complications, and death. Authored by Dr. Emily Riehm Meier, Pediatric Hematologist and Sickle Cell Research Director at the Indiana Hemophilia and Thrombosis Center in Indianapolis, these CME activities detail the most important and modifiable barriers to better outcomes among AYAs with SCD: underutilization of hydroxyurea, lack of comprehensive care for these patients, and need for structured transition programs to shepherd patients through the transition period. Would you like to gain more education about these important and timely topics - and earn free CMEs? https://partnersprn.org/online/index.php?category=0-25 These CME activities offer a patient-centered and practical approach to managing the care and transition of AYAs with SCD with a vision toward extending life expectancy, improving quality of life, and increasing competency among care providers for persons with SCD. Learn more by clicking the link. To complete an activity, you must be registered and logged in. https://partnersprn.org/online/index.php?category=0-25 Since 1983, the United States government has recognized September as Sickle Cell Awareness Month. In the thirty-four years since, a great deal of progress has been made with regards to research and treatment. In fact, activity in these two combined areas has led to a doubling of the average life expectancy for Sickle Cell patients in the United States. That is obviously great news that we can all appreciate and salute. We are making great strides at an accelerated rate and with the advent of the new CRISPR technology (https://www.yourgenome.org/facts/what-is-crispr-cas9), we stand on the verge of having a cure that, one day, can be accessible to more patients than stem cell transplants. But that day is still in the future.
The Sickle Cell community applauds these advances, but we need to remain focused on those individuals that face day after day of unrelenting pain, missed life opportunities and burdensome financial challenges. We must remain fixated on the needs of patients and families who face lives of uncertainty and recurrent flirtations with despair. And we should remain determined to do everything in our power to mitigate and/or minimize the many compounding issues that affect the members of our Sickle Cell community. We are not alone in this and we remain grateful that, now more than ever before, Sickle Cell advocacy and activism is at an all-time high. This brings us to an important point. For thirty-four years we have heard the words “Sickle Cell Awareness.” Awareness is only one part of the solution. Even so, it continues to amaze us that we still have so much awareness work to do. Too many people still don’t know much about Sickle Cell. A nursing student recently said, “I wish they would have spent more time on Sickle Cell. One class. That’s all we had.” This young woman was aware of Sickle Cell but she really didn’t know how to care for people who have it. This is just one example of why we need more than awareness to make a meaningful impact on this illness. We need to combine increased awareness with increased action. Whether it is institutionalizing a way that ensures that healthcare providers undergo more intensive training on Sickle Cell care or creating increased opportunities for Sickle Cell patients to access higher levels of education and employment, we need to take action that can make a difference. Awareness can’t do that by itself. It takes action to make things change. Gary Gibson; President/CEO Martin Center Sickle Cell Initiative How important is community connection? Are there benefits to feeling like you're part of a group? The Indiana Sickle Cell Consortium (ISCC) believes that a sense of community is important for everyone’s wellbeing, especially those with a chronic condition. Connecting with other people who can relate to the ups and downs of living with a chronic condition help ensure you don’t feel like you’re alone. That's why consortium members work to connect those affected by sickle cell to others with similar experiences.
Our most recent community outreach project involved ISCC booth representation at the INShape Indiana Black and Minority Health Fair from July 13th to 16th. At this event we had the opportunity to talk to over 1,000 people about sickle cell disease and the importance of getting tested for sickle cell trait. Several people with sickle cell disease volunteered to staff our booth, and this was a chance for them to talk about their experiences with health fair attendees. What’s the next event in the pipeline? The 9th Annual Sickle Cell-abration on September 30, 2017! (Check out event details here: https://2017sicklecellabration.eventbrite.com). This event, which includes education about sickle cell disease, will also feature a fashion show. Fashion show models will all be people with sickle cell disease. The fashion show will be a chance for patients to share their experiences and meet other people who are living with or affected by sickle cell disease. Feeling part of a larger community is important for those who are living with a chronic illness. The ISCC is striving to help people living with sickle cell create that community for themselves. If you're interested in volunteering or participating in any of our events contact the consortium at ISCC@ihtc.org. Ellen Bloom, MPH, CHES; Sickle SAFE Program Coordinator The Indiana Hemophilia & Thrombosis Center Written By: Dr. Emily Meier, Pediatric Hematologist and Sarah Hall, MHS, PA-C of Indiana Hemophilia and Thrombosis Center, Inc.
|